Inventio
Vol. 19, núm. 49, 2023
doi: http://doi.org/inventio/10.30973/2023.19.49/8
La odisea de las enfermedades raras
The odyssey of rare diseases
Gabriela Denisse Mata Salgado
denisse.mata.salgado@gmail.com
Centro de Investigación en Dinámica Celular (cidc), Universidad Autónoma del Estado de Morelos (uaem)
Iván Martínez-Duncker Ramírez
orcid: 0000-0001-5626-2892, duncker@uaem.mx
Centro de Investigación en Dinámica Celular (cidc), Universidad Autónoma del Estado de Morelos (uaem)
Mario Ernesto Cruz Muñoz
orcid: 0000-0001-6851-708X, mario.cruz@uaem.mx
Facultad de Medicina (fm), Universidad Autónoma del Estado de Morelos (uaem)
resumen
Las personas afectadas por enfermedades raras se enfrentan a desafíos únicos que les limitan el obtener un diagnóstico rápido, sufriendo así lo que se conoce como odisea diagnóstica. En este artículo se comentan los principales elementos que condicionan esta odisea y las estrategias para enfrentarla. De igual forma, se describen algunas de las principales estrategias y tecnologías desarrolladas para aportar tratamientos correctivos a quienes sufren de enfermedades raras, ya que la mayor parte de los más de siete mil tipos de enfermedades raras no cuentan con un tratamiento correctivo, lo cual afecta el acceso de las personas afectadas a una mejor calidad de vida.
palabras clave
enfermedades raras, diagnóstico genético, secuenciación de nueva generación, terapia génica, terapia celular
abstract
People affected by rare diseases face unique challenges that limit them from obtaining a rapid diagnosis, thus suffering from what is known as a diagnostic odyssey. This article discusses the main elements that condition this odyssey and the strategies to face it. Likewise, some of the main strategies and technologies developed to provide corrective treatments to those suffering from rare diseases are described, considering that most of the more than seven thousand types of rare diseases do not have corrective treatment, thus affecting the access of affected people to a better quality of life.
key words
rare diseases, genetic diagnosis, next generation sequencing, gene therapy, cell therapy
Recepción: 29/08/23. Aceptación: 23/02/24. Publicación: 21/05/24.
Introducción
El diagnóstico y tratamiento de las enfermedades más comunes acapara gran parte de la atención médica y científica, así como de la sociedad en general; sin embargo, existen otras enfermedades menos frecuentes llamadas enfermedades raras, consideradas tanto por la Organización Mundial de la Salud (oms) como por el gobierno de México, como aquellas que afectan a menos de cinco personas por cada diez mil habitantes. Parte del problema de padecer este tipo de enfermedades es que los médicos no están acostumbrados a identificarlas, lo que lleva a desconocer cómo se manifiestan, pero también a confundirlas con otras enfermedades y establecer diagnósticos equivocados.
Adicionalmente, aun cuando el médico las sospeche, no siempre hay acceso a las tecnologías que permiten diagnosticarlas y, lo que es aún más grave, aproximadamente el 95% de las enfermedades raras carece de un tratamiento específico (figura 1). Estas circunstancias, entre otros retos, llevan a las personas afectadas por enfermedades raras, así como a sus familias, a enfrentar situaciones muy difíciles. Por ello sus historias son de lucha, resiliencia y esperanza (Danese y Lippi, 2018; Haendel et al., 2020; Pletcher et al., 2007).
Es sorprendente que las enfermedades raras no sean tan raras como se percibe. Si sumamos a todas las personas afectadas por los más de siete mil tipos de enfermedades raras que se han identificado, se estima que existen cuatrocientos millones de personas afectadas a nivel global. Así que, aunque cada enfermedad rara es poco común, en conjunto representan un problema de salud pública mundial muy importante (Haendel et al., 2020; Roth y Marson, 2022).
Entre los desafíos que presenta el manejo de las enfermedades raras se encuentra que pueden manifestarse en cualquier etapa de la vida, si bien en general ocurren en los primeros años de vida (50-70%), y en muchos casos se trata de enfermedades que empeoran conforme pasa el tiempo (figura 1). Además, se acompañan con frecuencia de una mortalidad importante: aproximadamente un 26% de los afectados por una enfermedad rara mueren antes de los cinco años de edad. Todo lo anterior genera una carga física y emocional abrumadora tanto para las personas afectadas como para sus familias (Bavisetty et al., 2013; Marimpietri y Zuccari, 2023; The Lancet Global Health, 2024).
Alrededor de un 80% de las enfermedades raras son de origen genético, es decir, son causadas por alteraciones en los genes, llamadas mutaciones o variantes patogénicas (cambios malignos). Los genes son unidades de información codificada en nuestro adn (ácido desoxirribonucleico) que contienen las instrucciones para producir una proteína en particular. Las proteínas son las macromoléculas responsables de la mayor parte de las funciones que cumple una célula. Por ello, cuando un gen está afectado por una variante patogénica esto significa que la proteína para la que ese gen codifica estará alterada de tal forma que no podrá realizar su función adecuadamente, lo que implica que habrá procesos celulares afectados que eventualmente causarán una enfermedad (Bavisetty et al., 2013; The Lancet Global Health, 2024).
Dado que la mayoría de las enfermedades raras son genéticas, resulta crucial establecer un diagnóstico de este tipo, es decir, conocer con precisión qué gen o genes presentan variantes patogénicas. Esto casi siempre permite saber cuál es el mejor tratamiento conocido para una enfermedad genética. Lamentablemente, muchas personas afectadas por enfermedades raras tardan mucho en obtener un diagnóstico genético o incluso nunca lo logran, con lo cual se ven impedidas de recibir los potenciales beneficios (Marimpietri y Zuccari, 2023).
Figura 1
Impacto global de las enfermedades raras
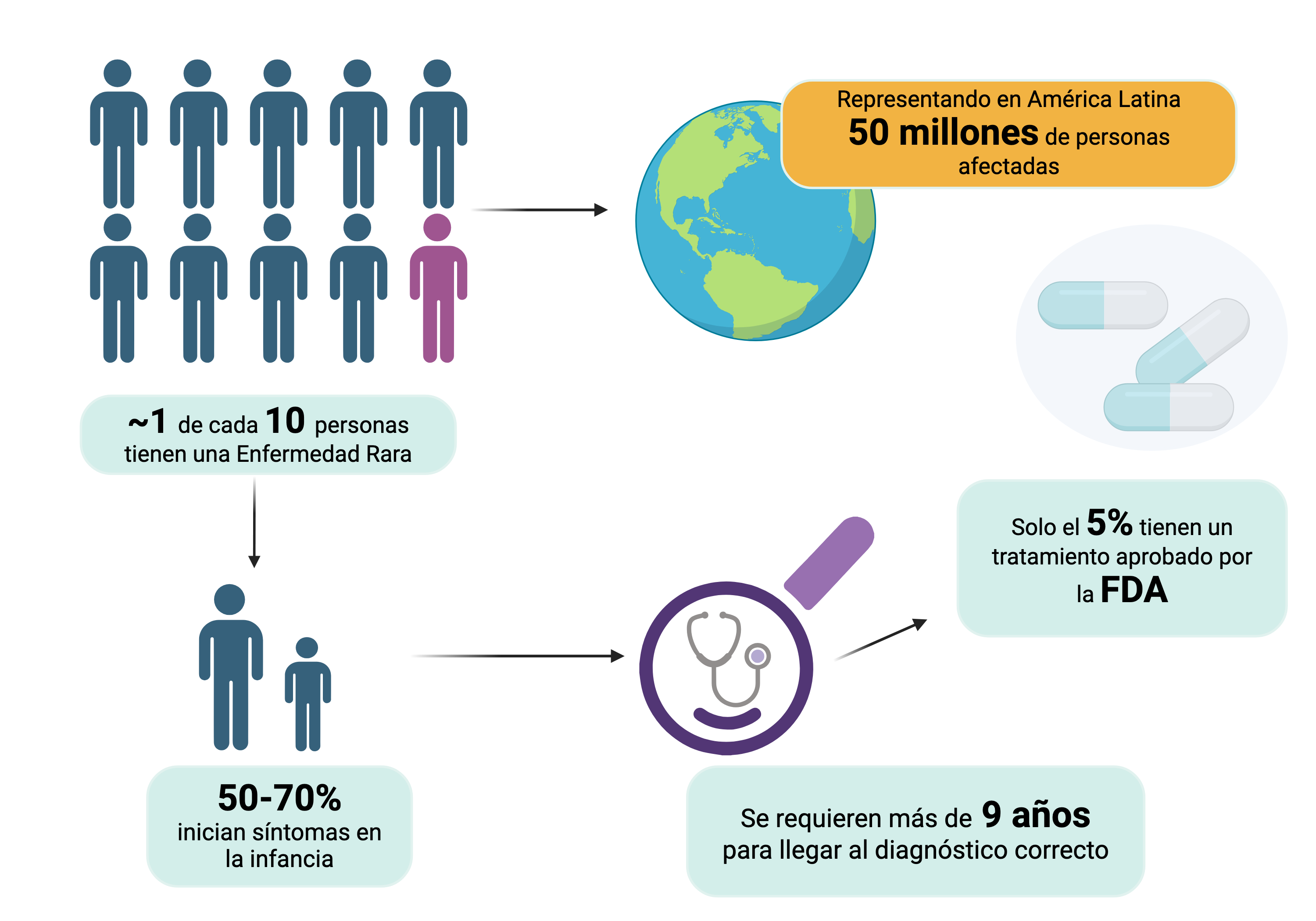
Aproximadamente, una de cada diez personas en todo el mundo sufre de una enfermedad rara, lo que equivale a unos cincuenta millones de personas afectadas en América Latina. De forma sorprendente, la mayoría de las personas que padecen alguna de estas enfermedades experimentan síntomas desde la infancia. Sin embargo, a pesar de esta realidad, se necesita de más de nueve años en promedio para obtener un diagnóstico preciso. Además del desafío diagnóstico, existe otro obstáculo aún mayor: la falta de opciones de tratamiento específicas para estas condiciones médicas.
Fuente: elaboración propia.
La odisea diagnóstica
El camino para obtener un diagnóstico genético de una enfermedad rara puede ser una odisea, palabra que proviene del título del poema homérico que describe las aventuras de Odiseo, también conocido como Ulises, en su largo viaje de regreso a su patria, Ítaca. Así, las personas afectadas y sus familias pasan por infinidad de consultas médicas y pruebas exhaustivas que, en muchas ocasiones, no conducen a un diagnóstico preciso. La demora en la obtención de este diagnóstico retrasa el inicio de un tratamiento adecuado. Desafortunadamente, en muchos casos, si no se actúa de manera oportuna, la muerte puede llegar antes del diagnóstico (Bauskis et al., 2022; Marimpietri y Zuccari, 2023).
Es importante considerar que las familias también sufren de esta odisea, la cual puede tener un impacto grave en la salud mental de quienes forman parte de ella. La falta de diagnóstico, las consultas frecuentes y el ver que los médicos dan tratamientos pero sin saber el diagnóstico puede causar ansiedad, frustración e impotencia. De igual forma, se sufre de un impacto financiero importante debido a los costos de las consultas, las pruebas diagnósticas y los días de trabajo perdidos que se acumulan rápidamente.
Acortando la odisea: ¿cómo se diagnostican las enfermedades raras?
Las enfermedades raras plantean un desafío para los médicos. Son afecciones poco comunes y diversas que requieren de un abordaje muy cuidadoso y de la colaboración de distintos expertos. Primero, los médicos deben recopilar la información detallada sobre los síntomas que experimentan las personas, o bien sobre los resultados que indican alguna anormalidad bioquímica o celular, así como identificar la presencia de casos similares en la familia de la persona afectada, presentes o pasados (antecedentes familiares). Cada detalle puede ser crucial para desentrañar el enigma (Hayward y Chitty, 2018, Roth y Marson, 2021).
A medida que los médicos exploran los síntomas del paciente pueden derivarlos a diferentes especialistas, quienes evalúan y examinan los aspectos específicos de la enfermedad para obtener una visión más completa. Frecuentemente se realizan pruebas de laboratorio o de imágenes, como radiografías, ultrasonidos, tomografías computarizadas y, en caso necesario, también se toman pequeñas muestras (biopsias) de alguno de los tejidos del cuerpo que se sospechan afectados, con la finalidad de obtener pistas valiosas que apunten a la causa subyacente. Por ejemplo, en el caso de enfermedades raras conocidas como distrofias musculares y que afectan la capacidad para caminar, se pueden tomar biopsias de los músculos tanto para estudiar la forma de las células musculares como para determinar si hay proteínas que les estén faltando para realizar su función de manera adecuada (figura 2a).
Una vez que se integran todos los datos necesarios y el médico tratante sospecha de una enfermedad de causa genética, el siguiente paso es derivar a los pacientes al médico genetista. La función del médico genetista es fundamental en el proceso de diagnóstico de enfermedades raras con base genética. Estos especialistas están capacitados en el estudio de los genes y su relación con las enfermedades (figura 2b).
Los médicos genetistas considerarán la realización de pruebas genéticas como parte integral del proceso diagnóstico. Estas pruebas analizan el material genético de la persona, generalmente un tipo de material llamado ácido desoxirribonucleico o adn. El lector puede imaginar el adn como una biblioteca compuesta por libros que contienen todas las instrucciones para el funcionamiento del cuerpo (Roth y Marson, 2021).
En primer lugar, se puede realizar un cariotipo, que es una técnica que examina la estructura general de los cromosomas para detectar alteraciones visibles. Siguiendo con la analogía anterior, sería como revisar que los libros de la biblioteca estén completos y en buenas condiciones; sin embargo, en muchos casos se requiere un análisis más detallado, por lo que se pueden solicitar estudios que analicen cada letra del libro genético, en la búsqueda de palabras faltantes, duplicadas o mal escritas, y que tengan como consecuencia una afectación a la función del gen. Como mencionamos antes, estas variaciones genéticas con respecto a la mayor parte de los individuos de una población, y que tienen el potencial de afectar la función del gen, se llaman variantes patogénicas. Para llevar a cabo este análisis minucioso se utilizan técnicas sofisticadas de secuenciación genética, es decir, tecnología que permite leer el adn (Marwaha et al., 2022).
La importancia de tener la comprobación diagnóstica con estas pruebas genéticas ayuda, primero, a los pacientes y a sus familias a, finalmente, entender la causa de la enfermedad, una interrogante que desgasta mucho a los padres. En segundo lugar, les permite acceder a información sobre el pronóstico de la enfermedad, es decir, qué va a ocurrir en los próximos meses o años, las implicaciones para la familia y las opciones de manejo y tratamiento disponibles para tomar decisiones informadas y recibir apoyo adecuado.
Figura 2a
Evaluación médica inicial y su papel en el diagnóstico genético
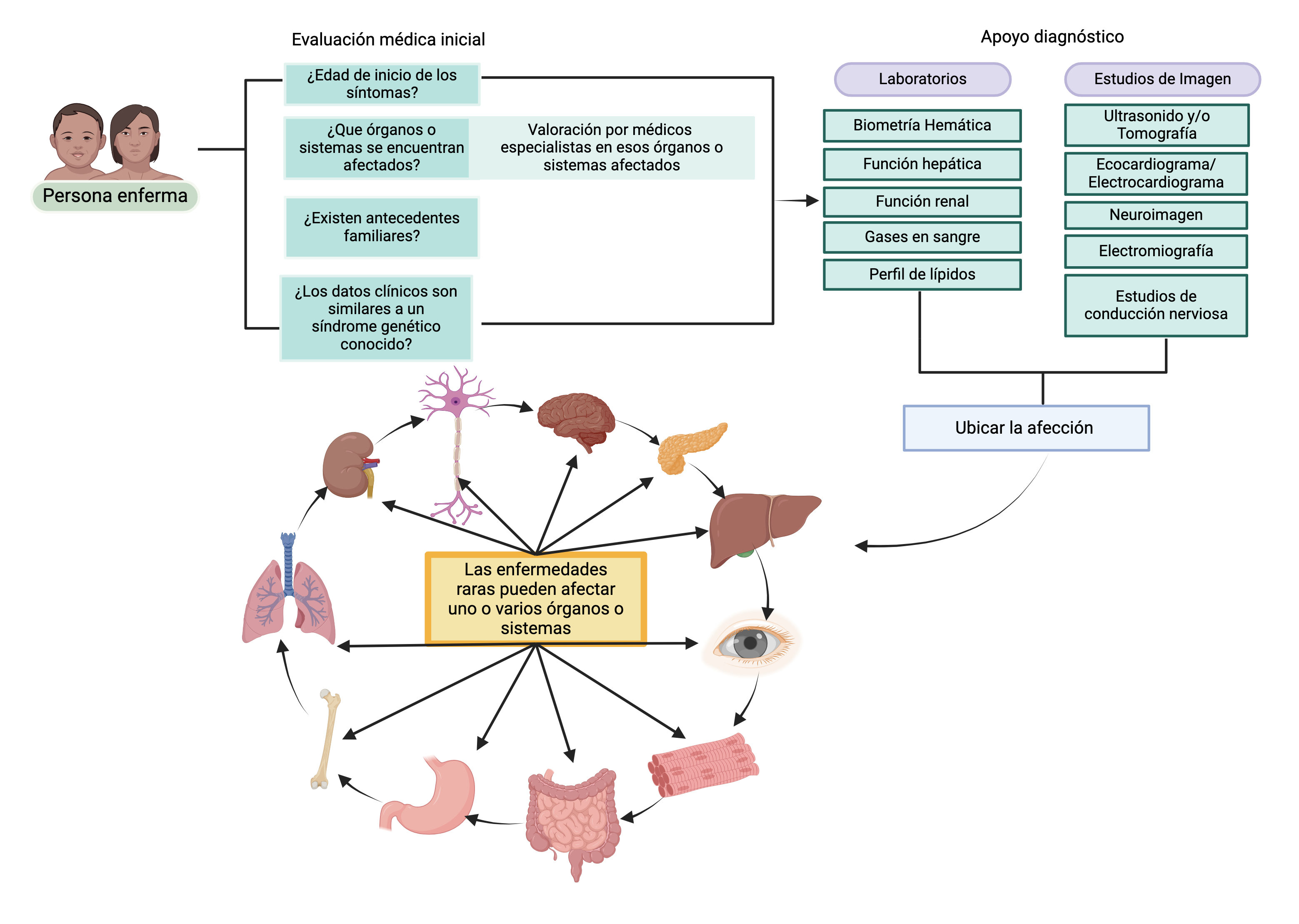
La evaluación médica inicial desempeña un papel fundamental en la orientación hacia un diagnóstico genético preciso. Mediante la formulación de preguntas dirigidas y la utilización de apoyos diagnósticos, como estudios de laboratorio y estudios de imagen, se sientan las bases para alcanzar un diagnóstico genético certero. La colaboración con un médico genetista se torna esencial en este proceso de integración experta.
Fuente: elaboración propia.
Figura 2b
El papel de los médicos genetistas en el proceso diagnóstico
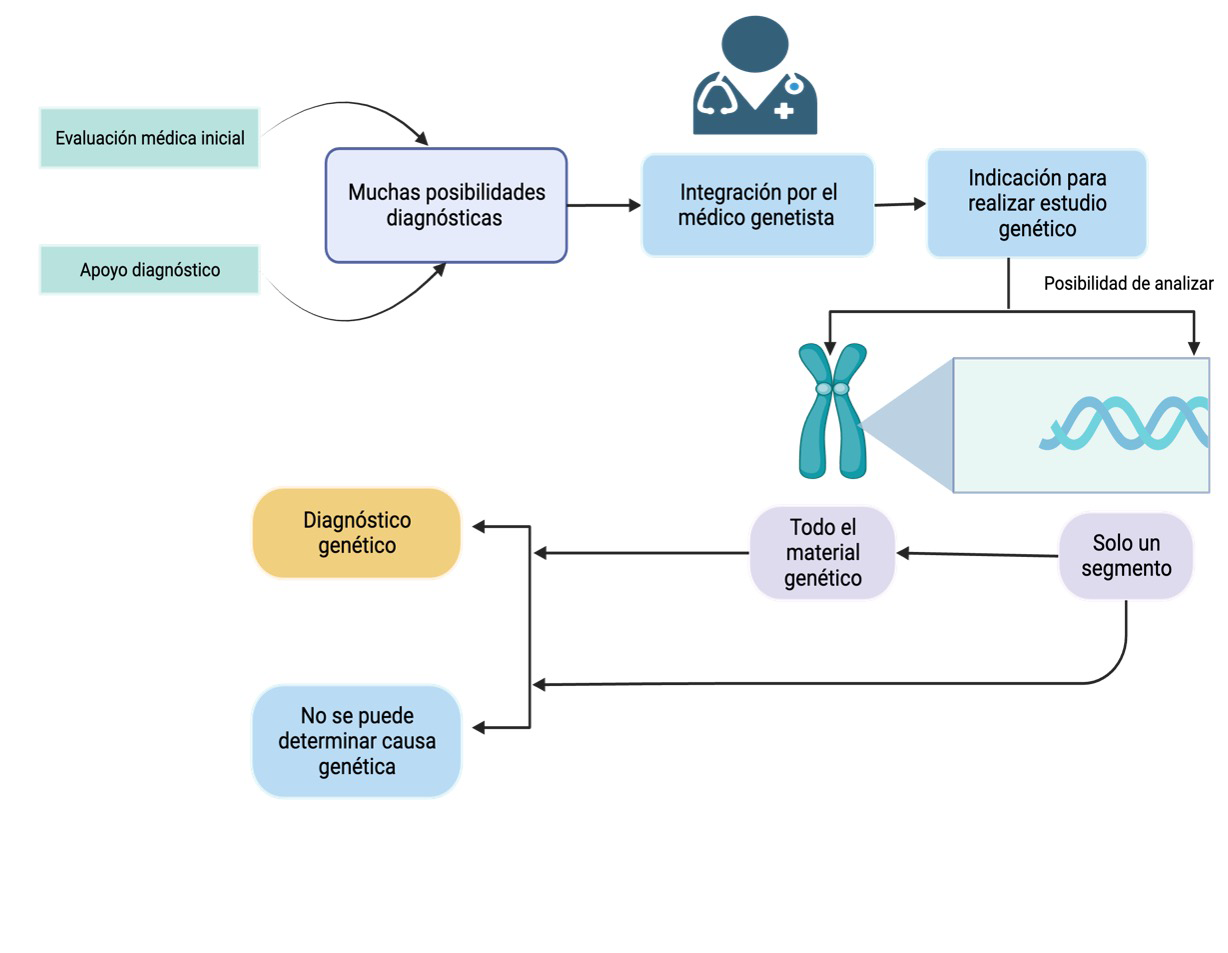
Los médicos genetistas desempeñan un papel central en reunir datos clínicos, antecedentes personales y familiares, así como en revisar estudios de laboratorio e imágenes médicas para formular un posible diagnóstico. Para confirmar este diagnóstico y profundizar en él se pueden llevar a cabo estudios genéticos que analizan segmentos específicos o la totalidad del material genético del paciente. Estos estudios genéticos proporcionan información esencial para identificar las causas subyacentes de las enfermedades raras y mejorar la precisión diagnóstica.
Fuente: elaboración propia.
Innovando el futuro: tecnologías novedosas para el diagnóstico genético
Una de las tecnologías más revolucionarias en el diagnóstico genético es la secuenciación de nueva generación (ngs, por sus siglas en inglés). Esta técnica ha transformado la forma en que estudiamos los genes y ha hecho posible analizar grandes cantidades de información genética de manera rápida y asequible. Con la ngs, los científicos pueden leer (secuenciar) no sólo un gen específico (segmento de material genético), sino el material genético completo de una persona.
Esto proporciona una visión detallada de todas las variantes genéticas presentes y ayuda a identificar las variantes patogénicas responsables de enfermedades raras. Dentro de esta tecnología destaca el exoma clínico, que se enfoca en regiones del libro genético que contiene las instrucciones para construir proteínas, las cuales funcionan como piezas indispensables para el buen funcionamiento del cuerpo. El exoma clínico se enfoca en estas secciones cruciales, donde a menudo se encuentran cambios en individuos con ciertos problemas médicos (Roth y Marson, 2021).
Por otra parte, la inteligencia artificial (ia) ha comenzado a desempeñar un papel crucial en el diagnóstico genético. Mediante algoritmos de aprendizaje automático, la ia puede examinar conjuntos de datos genéticos y descubrir patrones que podrían escapar al ojo humano. ¿Qué son estos patrones? El lector puede imaginarse una especie de huella genética que se repite en diferentes personas que presentan una cierta enfermedad. Estos patrones revelan conexiones sutiles que podrían desencadenar una afección. Aquí es donde entra en juego el algoritmo de aprendizaje automático, que es una especie de cerebro electrónico entrenado para identificar y comprender estos patrones (Wojtara et al., 2023).
La ia no sólo cumple un papel detectando patrones, sino que también acelera el proceso de interpretar los resultados genéticos. Ahora el lector podría imaginarse que debe armar un gran rompecabezas de datos genéticos; la ia sería como una mente experta que puede ensamblar rápidamente las piezas correctas. Además, la ia amplifica la precisión del diagnóstico, ya que su capacidad para analizar datos a una velocidad asombrosa y considerar numerosos factores en ello puede llevar a conclusiones más certeras (Wojtara et al., 2023).
Estas tecnologías novedosas no sólo están transformando el campo del diagnóstico genético, sino que también brindan esperanza a las personas con enfermedades raras y sus familias. Con diagnósticos más precisos, los pacientes pueden recibir tratamientos más adecuados y personalizados, lo que mejora su calidad de vida y les da un mayor control sobre su salud.
No podemos pasar por alto que estas tecnologías también nos plantean retos éticos y legales. La seguridad de nuestros datos genéticos y las preocupaciones sobre cómo podrían usarse de manera indebida son temas que necesitamos manejar con mucho cuidado mientras avanzamos en esta era en la que la investigación gira en torno a los genes, su funcionamiento, su interacción y su relación con las enfermedades.
En busca de una cura
La ausencia de tratamientos efectivos para las enfermedades raras plantea un desafío apremiante en el ámbito de la salud. Con frecuencia, estas afecciones son complejas y poco comprendidas, lo que dificulta el desarrollo de terapias específicas, aunado a que las grandes corporaciones farmacéuticas no se deciden a invertir en el desarrollo de tratamientos para enfermedades que afectan a millones de personas. Esta falta de opciones terapéuticas incide en la calidad de vida de quienes las padecen. La falta de tratamientos adecuados para el conjunto de enfermedades raras resalta la urgencia de impulsar investigaciones y desarrollos médicos orientados a abordar estas condiciones de manera más precisa y brindar soluciones tangibles a quienes enfrentan estas realidades desafiantes (Roth y Marson, 2021).
A pesar de los retos mencionados, la terapia de reemplazo enzimático ha demostrado ser una aplicación exitosa, en particular en el caso de enfermedades lisosomales. Estas condiciones se caracterizan por deficiencias en las enzimas encargadas de degradar componentes celulares en los lisosomas. La ingeniería genética ha proporcionado una solución a este problema, al permitir la producción de estas enzimas en entornos de laboratorio, que luego se administran a los pacientes a través de infusiones intravenosas para corregir el defecto enzimático.
En la enfermedad de Fabry, donde hay una deficiencia de la enzima alfa-galactosidasa, se suministra la versión recombinante de ésta para ayudar a descomponer las moléculas acumuladas. Dos elementos fueron clave para el desarrollo de este tratamiento: encontrar la forma de producir suficientes cantidades de proteína recombinante y descubrir los mecanismos por los cuales la proteína, una vez en el torrente sanguíneo, podría ser dirigida específicamente a los lisosomas para realizar su función. La película Medidas extraordinarias (Sher et al., 2010) nos presenta un buen ejemplo de este proceso de innovación, para que el lector conozca más sobre él (Germain et al., 2019).
Otra aplicación exitosa es la terapia génica, que emerge como una de las áreas más prometedoras para el desarrollo de tratamientos innovadores. Su objetivo principal es corregir los defectos genéticos subyacentes que provocan las enfermedades hereditarias. Esta técnica implica la introducción de genes sanos en las células afectadas para el reemplazo de los genes afectados. Con la terapia génica se abre la posibilidad real de curar enfermedades genéticas raras y, en un futuro no muy lejano, también algunos trastornos más comunes (Gonçalves y Alves, 2017).
Un ejemplo importante de terapia génica es el medicamento Zolgensma, que ha sido aprobado para tratar una enfermedad llamada atrofia muscular espinal (sma, por sus siglas en inglés). La sma es una condición genética que causa debilidad muscular progresiva y parálisis debido a mutaciones en el gen smn1, que provoca la pérdida de producción de una proteína esencial para el funcionamiento de las neuronas motoras, las cuales son las encargadas de controlar el movimiento de los músculos (Ogbonmide et al., 2023).
Zolgensma se administra mediante una infusión intravenosa en niños menores de dos años. El medicamento utiliza un virus modificado genéticamente, llamado virus adenoasociado (aav), para entregar una copia funcional del gen smn1 a las células del sistema nervioso central. Una vez dentro de las células, el gen funcional del smn1 produce la proteína smn, necesaria para la supervivencia y el funcionamiento adecuado de las neuronas motoras, lo cual revierte los síntomas. Esta terapia génica ha demostrado ser efectiva en ensayos clínicos y ha brindado esperanza a las familias afectadas por esta enfermedad (Day et al., 2022).
Existen otras estrategias, como la terapia celular, que buscan reemplazar las células enfermas por células sanas. Por ejemplo, en la anemia de Fanconi, una enfermedad genética rara que afecta la producción de células sanguíneas, esta técnica se aplica para reemplazar las células productoras de células sanguíneas (células madre), afectadas por variantes patogénicas, por células madre sanas. Éstas pueden derivarse de una persona donadora, o bien desarrollarse a partir de las células del propio paciente, tomando células de su piel (llamadas fibroblastos), quitándoles la mutación y reprogramándolas para ser células madre (Roth y Marson, 2021; Sivandzade y Cucullo, 2021).
Por otra parte, hay tecnologías que permiten corregir la mutación en las células. Una de ellas es crispr, que permite editar genes de manera precisa y eficiente. Los componentes de esta tecnología, una vez introducidos en el cuerpo, van al sitio del adn que está afectado por una mutación genética responsable de enfermedades y la elimina de forma específica, ya que el diseño de los componentes incluye la información sobre el sitio de adn donde se requiere hacer la edición, lo cual la hace una tecnología muy flexible (Roth y Marson, 202; Uddin et al., 2020).
Desarrollar una molécula completamente nueva implica costos elevados, lo que despierta un limitado interés por parte de las compañías farmacéuticas en la investigación de nuevos medicamentos para tratar enfermedades raras. Como alternativa, familias y científicos han optado por colaborar para descubrir tratamientos efectivos mediante el aprovechamiento de medicamentos ya existentes, algo que se conoce como reposicionamiento farmacológico. Este enfoque, al no requerir la creación de moléculas nuevas, ha demostrado ser una estrategia más económica. Un ejemplo es el uso del fármaco Epalrestat, que ha sido empleado durante largo tiempo para tratar complicaciones vinculadas con la diabetes (Jonker et al., 2024).
En el contexto de la enfermedad rara pmm2-cdg, causada por mutaciones en la enzima pmm2 responsable de la producción de glicanos, los cuales son esenciales para la comunicación biológica, los científicos realizaron experimentos en pequeños frascos donde colocaron células de pacientes en contacto con miles de medicamentos, y descubrieron que el Epalrestat mejoraba la producción de glicanos en células afectadas por las mutaciones en el gen pmm2. Aunque en ese momento desconocían la causa de esta mejora, el resultado fue sumamente intrigante (Iyer et al., 2019).
Estudios posteriores lograron identificar que el Epalrestat activa la enzima pmm2, lo que contribuye a mejorar su función. Este hallazgo no sólo destaca la eficacia del reposicionamiento de fármacos en la investigación de tratamientos para enfermedades raras, sino que también revela cómo los compuestos ya existentes pueden ser redescubiertos y aplicados de manera innovadora en el ámbito médico (figura 3).
Figura 3
Avances terapéuticos innovadores
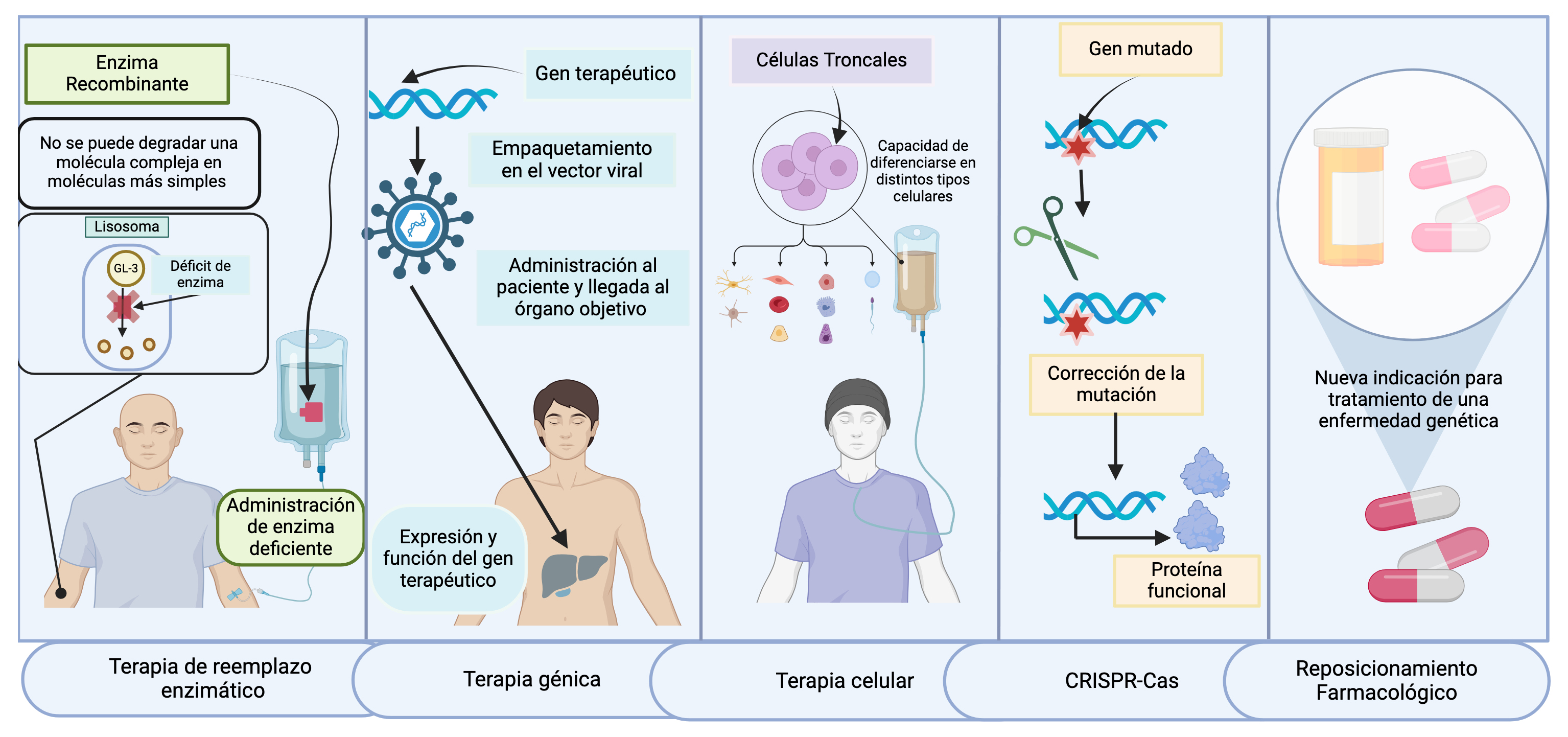
Se trata de tecnologías de vanguardia que están revolucionando la atención médica, al abarcar desde terapias de reemplazo enzimático y terapias génicas hasta terapias celulares, crispr-Cas y reposicionamiento farmacológico. Estas tecnologías innovadoras están redefiniendo las estrategias de tratamiento médico y abriendo nuevas perspectivas hacia una medicina más personalizada y efectiva.
Fuente: elaboración propia.
Conclusiones
Las enfermedades raras plantean un desafío sustancial tanto en el ámbito médico como en el social, que va desde lo diagnóstico hasta la accesibilidad a tratamientos. Aunque se les califique como raras, su impacto trasciende, pues dejan una profunda huella en las vidas de quienes las padecen y en sus familias. No obstante, hemos avanzado gracias a la tecnología para acortar la odisea diagnóstica y mejorar la oferta tecnológica para generar tratamientos que permitan una mejor calidad de vida. Esta tarea es una labor que nos debe interesar a la sociedad como conjunto, pues requiere enfrentar desafíos ante un sistema de salud y económico que margina a quienes sufren este tipo de enfermedades.
Referencias
Bauskis, A., Strange, C., Molster, C. y Fisher, C. (2022). The diagnostic odyssey: insights from parents of children living with an undiagnosed condition. Orphanet Journal of Rare Diseases, 17(1), 233. https://ojrd.biomedcentral.com/articles/10.1186/s13023-022-02358-x
Bavisetty, S., Grody, W. W. y Yazdani, S. (2013). Emergence of pediatric rare diseases: review of present policies and opportunities for improvement. Rare Diseases, 1(1), e23579. https://doi.org/10.4161/rdis.23579
Danese, E. y Lippi, G. (2018). Rare diseases: the paradox of an emerging challenge. Annals of Translational Medicine, 6(17), sp. https://doi.org/10.21037%2Fatm.2018.09.04
Day, J. W., Howell, K., Place, A., Long, K., Rossello, J., Kertesz, N. y Nomikos, G. (2022). Advances and limitations for the treatment of spinal muscular atrophy. bmc Pediatrics, 22(1), 632. https://doi.org/10.1186/s12887-022-03671-x
Germain, D. P., Elliott, P. M., Falissard, B., Fomin, V. V., Hilz, M. J., Jovanovic, A., Kantola, I., Linhart, A., Mignani, R., Namdar, M., Nowak, A., Oliveira, J.-P., Pieroni, M., Viana-Baptista, M., Wanner, C., y Spada, M. (2019). The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Molecular Genetics and Metabolism Reports, 19, 100454. https://doi.org/10.1016/j.ymgmr.2019.100454
Gonçalves, G. A. R. y Paiva, R. D. M. A. (2017). Gene therapy: advances, challenges and perspectives. Einstein (Sao Paulo), 15, 369-375. https://doi.org/10.1590%2FS1679-45082017RB4024
Haendel, M., Vasilevsky, N., Unni, D., Bologa, C., Harris, N., Rehm, H., Hamosh, A., Baynam, G., Groza, T., McMurry, J., Dawkins, H., Rath, A., Thaxton, C., Bocci, G., Joachimiak, Marcin P., Köhler, S., Robinson, P. N., Mungall, C. y Oprea, T. I. (2020). How many rare diseases are there? Nature Reviews Drug Discovery, 19(2), 77-78. https://doi.org/10.1038/d41573-019-00180-y
Hayward, J. y Chitty, L. S. (2018). Beyond screening for chromosomal abnormalities: advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing. Seminars in Fetal and Neonatal Medicine, 23(2), 94-101. https://doi.org/10.1016/j.siny.2017.12.002
Iyer, S., Sam, F. S., DiPrimio, N., Preston, G., Verheijen, J., Murthy, K., Parton, Z., Tsang, H., Lao, J., Morava, E. y Perlstein, E. O. (2019). Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation pmm2-cdg. Disease Models & Mechanisms, 12(11), dmm040584. https://doi.org/10.1242/dmm.040584
Jonker, A. H., O’Connor, D., Cavaller-Bellaubi, M., Fetro, C., Gogou, M., ’T Hoen, P. A. C., Kort, M., Stone, H., Valentine, N. y Pasmooij, A. M. G. (2024). Drug repurposing for rare: progress and opportunities for the rare disease community. Frontiers in Medicine, 11, 1352803. https://doi.org/10.3389/fmed.2024.1352803
Marimpietri, D. y Zuccari, G. (2023). Development of medicines for rare pediatric diseases. Pharmaceuticals, 16(4), 513. https://doi.org/10.3390%2Fph16040513
Marwaha, S., Knowles, J. W. y Ashley, E. A. (2022). A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Medicine, 14(1), 23. https://doi.org/10.1186/s13073-022-01026-w
Ogbonmide, T., Rathore, R., Rangrej, S. B., Hutchinson, S., Lewis, M., Ojilere, S., Carvalho, V. y Kelly, I. (2023). Gene therapy for spinal muscular atrophy (sma): a review of current challenges and safety considerations for onasemnogene abeparvovec (Zolgensma). Cureus, 15(3), sp. https://doi.org/10.7759/cureus.36197
Pletcher, B. A., Toriello, H. V., Noblin, S. J., Seaver, L. H., Driscoll, D. A., Bennett, R. L. y Gross, S. J. (2007). Indications for genetic referral: a guide for healthcare providers. Genetics in Medicine, 9(6), 385-389. https://doi.org/10.1097/gim.0b013e318064e70c
Roth, T. L. y Marson, A. (2021). Genetic disease and therapy. Annual Review of Pathology: Mechanisms of Disease, 16, 145-166. https://doi.org/10.1146/annurev-pathmechdis-012419-032626
Sher, S., Shamberg, M. (prods.) y Vaughan, T. (dir.) (2010). Medidas extraordinarias. Sony Pictures. 106 min.
Sivandzade, F. y Cucullo, L. (2021). Regenerative stem cell therapy for neurodegenerative diseases: an overview. International Journal of Molecular Sciences, 22(4), 2153. https://doi.org/10.3390/ijms22042153
The Lancet Global Health (2024). The landscape for rare diseases in 2024. The Lancet Global Health, 12(3), e341. https://doi.org/10.1016/s2214-109x(24)00056-1
Uddin, F., Rudin, C. M. y Sen, T. (2020). crispr gene therapy: applications, limitations, and implications for the future. Frontiers in Oncology, 10, 1387. https://doi.org/10.3389/fonc.2020.01387
Wojtara, M., Rana, E., Rahman, T., Khanna, P. y Singh, H. (2023). Artificial intelligence in rare disease diagnosis and treatment. Clinical and Translational Science, 16(11), 2106-2111. https://doi.org/10.1111/cts.13619